トップページ > 研究所組織 > 分子糖尿病医学研究部 > 研究紹介
研究紹介
2型糖尿病では、インスリン標的臓器(脂肪組織・筋肉・肝臓など)におけるインスリン抵抗性を背景として、膵β細胞での代償的なインスリン分泌増加の不全が生じています。 我々の研究部では、2型糖尿病発症に関わるインスリン標的臓器および膵β細胞を主な研究対象として、遺伝子改変マウスモデルや臨床検体を用い、個体における糖代謝恒常性の維持とその破綻のメカニズムを分子レベルで解析しています。そのゴールは、2型糖尿病の新たな治療戦略を提案していくことです。
主な研究プロジェクトは以下の通りです。
1. 肥満に起因するインスリン抵抗性・糖尿病の発症メカニズムの解明肥満とそれに関連する代謝性疾患は日本を含む先進国において最も重要な公衆衛生上の課題のひとつです。特に肥満によって誘導されるインスリン抵抗性は、2型糖尿病、循環器疾患および高血圧などを含む多数の代謝性疾患の増加に大きく関与していると考えられていることから、肥満を基盤としたインスリン抵抗性・糖尿病の予防法・新規治療法の開発を念頭に研究を行なっています。
インスリン抵抗性は、肥満の明確かつ中心的な特徴でありながら、その分子メカニズムの全容の詳細は明らかとなっていません。肥満は体脂肪が過剰に蓄積した状態であると定義されますが、肥満の脂肪組織では脂肪細胞の肥大化とともに、飽和脂肪酸や炎症性サイトカインが大量に産生および分泌されるようになる一方で、抗炎症性サイトカインの産生が減少するアディポサイトカイン調節異常を認めます。過剰に分泌された飽和脂肪酸が肝臓や骨格筋などのインスリン標的組織において異所性脂肪として蓄積するにとどまらず、炎症性サイトカインとともにシグナル分子として機能し、インスリン作用を阻害することによりインスリン感受性組織においてインスリン抵抗性を誘導します(図1)。これが、内臓脂肪型肥満を背景としてインスリン抵抗性を基盤とするメタボリックシンドロームの病態に深く関与すると考えられています。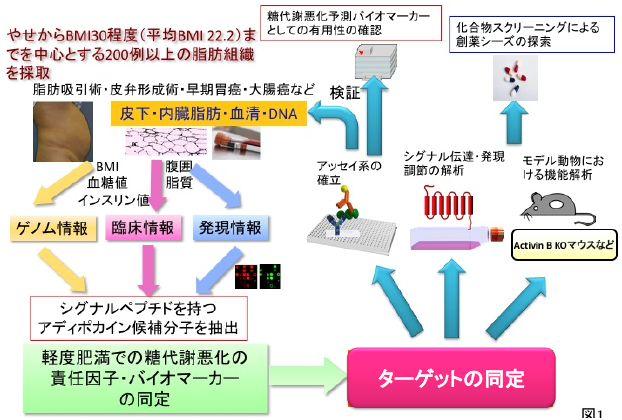
我々のグループでは、肥満脂肪組織における免疫細胞の浸潤が、肥満における炎症・インスリン抵抗性の発症原因ではないかとの仮説に基づいて検討しました。その結果、肥満マウスにおいてClass IB phosphatidylinositol 3-kinase (PI3K, PI3Kγ) を阻害することにより、脂肪組織のみならず肝臓における炎症を抑制し、全身のインスリン抵抗性が改善することを見出しました (Kobayashi N, et al. PNAS 2011)。
現在は、肥満に起因するインスリン抵抗性・糖尿病の発症を結びつける分子メカニズムを明らかにし、治療に応用するべく日々研究に取り組んでいます。
2. 膵β細胞の維持機構の解明と膵β細胞回復による2型糖尿病治療法の開発2型糖尿病は、インスリン標的臓器におけるインスリン抵抗性を背景として膵β細胞における代償的なインスリン分泌増加の不全が必発することから、膵β細胞の維持・破綻に関与するシグナルについて理解し、膵β細胞の機能的・量的な維持と回復をはかることは糖尿病の根治治療法開発と予防の鍵となります。
我々のグループでは、以前、膵β細胞特異的インスリン受容体 (IR) /Insulin-like growth factor-1 受容体 (IGF-1R) 欠損マウスの解析を行い、このマウスでは膵β細胞が重度に障害され、生後早期に重篤な糖尿病を発症することを見出し、膵β細胞でのインスリン作用の低下が、インスリン分泌の低下につながることを示しました (Ueki, et al. Nat. Genet. 2006)。これは、膵β細胞がインスリン分泌臓器であると同時に、インスリン作用臓器でもあることを示しています。
つづいて、膵β細胞におけるインスリンシグナルの作用についてさらに詳細に検討するため、インスリンシグナルの下流で活性化される主要な経路であるclass 1A phosphatidylinositol 3-kinase (PI3K)/Akt経路を膵β細胞で特異的に障害したマウス (RIPCre+/pik3r1f/f/pik3r2ko/koマウス, 以下βDKOマウス) の解析を行いました。このβDKOマウスでは、通常食条件下の成獣においてインスリン分泌の低下を伴う耐糖能異常が観察されました。しかし、膵島の形態や量はコントロールマウスに比べ明らかな差異を認めませんでした。そこで2光子顕微鏡を用い、グルコース応答性インスリン分泌の詳細な検討を進めたところ、βDKOマウス膵島の膵β細胞では、グルコース刺激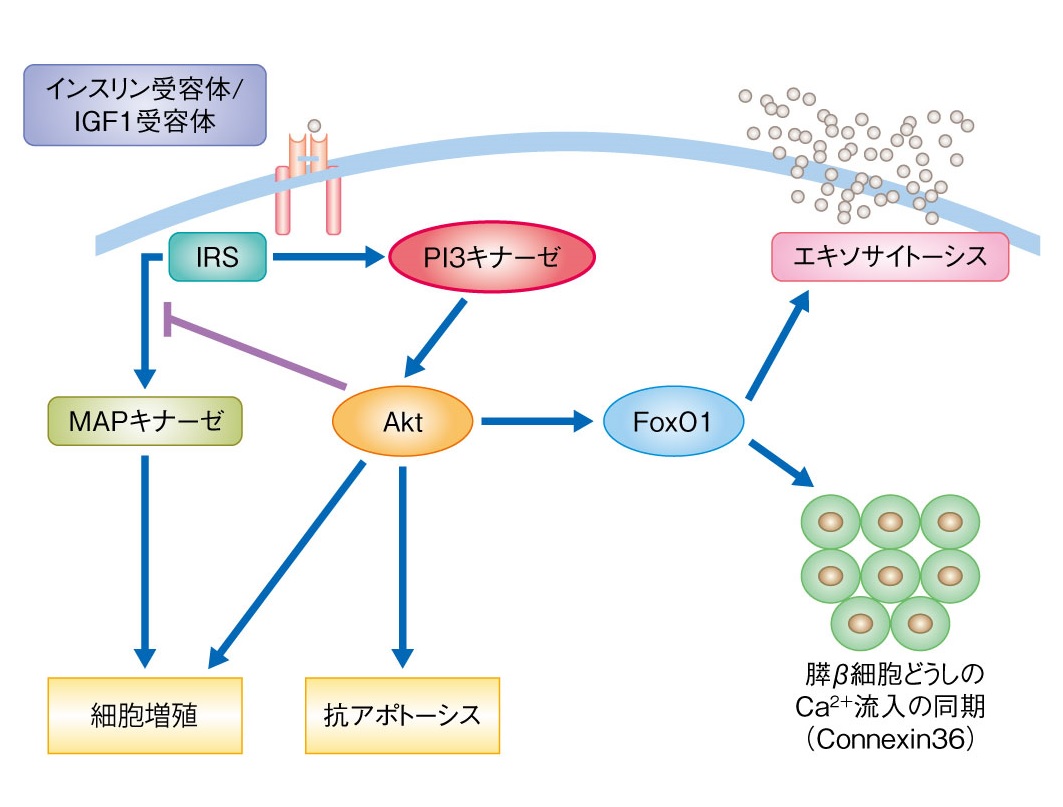 によるインスリン分泌顆粒の開口放出数が有意に減少しており、またグルコース刺激による細胞内へのCa2+流入の際の膵β細胞同士の同期にも障害が認められました。さらにケージドCa2+刺激による実験でも、インスリン分泌顆粒の開口放出の障害が観察されたことから、グルコース刺激による細胞内へのCa2+流入の際の膵β細胞同士の同期と、細胞内へのCa2+流入以降のインスリン分泌顆粒の開口放出の2段階で障害が生じていることが示唆されました。
によるインスリン分泌顆粒の開口放出数が有意に減少しており、またグルコース刺激による細胞内へのCa2+流入の際の膵β細胞同士の同期にも障害が認められました。さらにケージドCa2+刺激による実験でも、インスリン分泌顆粒の開口放出の障害が観察されたことから、グルコース刺激による細胞内へのCa2+流入の際の膵β細胞同士の同期と、細胞内へのCa2+流入以降のインスリン分泌顆粒の開口放出の2段階で障害が生じていることが示唆されました。
この原因について検討をすすめたところ、βDKOマウスの膵島ではインスリン分泌顆粒の開口放出を制御することが知られているsoluble N-ethylmaleimide attachment protein receptor (SNARE) タンパク質 (Syntaxin1A, Snap25等) の遺伝子発現・タンパクレベルが低下しており、また、β細胞間のCa2+流入の同期に重要なギャップジャンクションを構成するConnexin36の発現低下・タンパクレベルの低下も認められました。そこで、βDKOマウスの膵島に恒常活性型Aktを発現させたところ、これらSNAREタンパク質およびConnexin36の発現は増化し、グルコース応答性インスリン分泌も回復しました。さらに私たちは、これら遺伝子の発現の少なくとも一部はPI3K/AktシグナルによるFoxo1の制御を介して調節されている可能性を示し、以上から、PI3K/Aktシグナルが、細胞内へのCa2+流入の際の膵β細胞間の同期およびインスリン分泌顆粒の開口放出を担う遺伝子の発現を制御し、インスリン分泌を保っていることを明らかにしました (Kaneko, et al. Cell Metabol. 2010)。
現在は、こうしたこれまでの研究成果を元に、膵β細胞の量や機能の維持に重要なシグナルの包括的な理解に向け、更なる解析を進めています。こうした研究成果を膵β細胞の数・機能の維持・回復による新たな2型糖尿病治療戦略へと繋げることが私たちの目標です。
3. 尿病合併NASHにおける肝発癌の機序の解明糖尿病の治療の目標は、糖尿病症状を除くことはもとより、糖尿病に特徴的な合併症、糖尿病に併発しやすい合併症の発症、増悪を防ぎ、健康な人と同様な日常生活の質(QOL)を保ち、健康な人と変わらない寿命を全うすることにあります。
糖尿病の患者さんのQOLや寿命を損なう慢性合併症として、糖尿病の患者さんでは細小血管障害(網膜症、腎症、神経障害)、大血管障害(冠動脈疾患、脳卒中、末梢動脈疾患)のリスクが高いことが広く知られていましたが、最近の疫学研究により糖尿病の患者さんでは癌リスクも高くなっていることが明らかになり、中でも肝臓癌のリスクは比較的顕著に上昇していることが分かりました (Kasuga, Ueki, et al. Cancer Sci 2013)。
そもそも正常な肝臓は血糖調節を行う主要な臓器のひとつであり、いわゆるメタボリックシンドロームのような過栄養状態で脂肪蓄積をきたすと(これを脂肪肝といいます)、糖尿病を発症してしまうことがあります。また、脂肪肝の患者さんの一部では、肝臓に線維化をきたし(これを非アルコール性脂肪性肝炎:nonalcoholic steatohepatitis, NASHといいます)、肝硬変、肝発癌をきたすことが知られています。
このように糖尿病とNASH肝癌は共通の基盤的病態のもとで発症している可能性があり、多くのNASH肝癌の患者さんは糖尿病を合併しています。糖尿病は肝臓などの標的臓器におけるインスリン作用不足による高血糖を主徴としますが、糖尿病状態における血中インスリン濃度や血糖値の変化がどのようにNASH肝癌の発症に影響するか、その詳細は明らかにされていません。
私たちはこれらを明らかにし、糖尿病合併脂肪肝や糖尿病合併NASHの患者さんの治療法選択や、糖尿病合併NASH肝癌の新しい治療法や予防法の開発に役立てることを目標にしています。
統合生理学研究室(室長: 粟澤 元晴)
最新のゲノム解析から得られた知見をいかに代謝研究に応用するか
インスリン抵抗性メカニズムの解明は、その多因子的な成り立ちのために困難を極め、未だ全体像の把握には程遠い状態です。一方、近年のゲノム解析手法の進歩により、我々のゲノムの90%以上にも及ぶ領域が、何らかのレベルで転写されRNAを産出している、という驚くべき事実が明らかとなっています(ENCODE他)。
これらRNAの大半は非コードRNA (non-coding RNA: ncRNA)に分類されていますが、ではこのようなRNA分子は、我々の病態生理にどのような役割を果たしているのでしょうか。当研究室では新たに同定されたこれらncRNAについての知見を、肥満、インスリン抵抗性メカニズムの解明に役立てようと試みています。
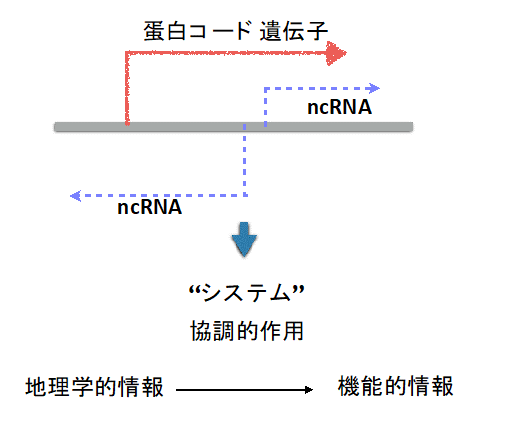
一般にncRNAの機能解析は容易ではなく、多くの場合我々の理解は、それらRNAがどこにあるか、という“地理学的”なレベルにとどまっています。これはncRNAの機能が多岐にわたり、かつ塩基配列からの機能を予測するメソッドも確立していないためで、アプローチには新たな視点が必要となります。さて転写がゲノムの広範にわたって起こるということの必然の結果として、一つの遺伝子座から複数の転写産物が産出されることがあります(Multiple-Coding、図3)。
このような場合興味深いことに、それら転写産物はしばしば互いに協調的に発現調節され、また共通の経路に作用することで“システム”として強い作用を発揮する可能性があります(Awazawa M, et al, Nat Med, 2017)。当研究室ではこの現象に注目することで、ncRNAの関わる新たな疾患メカニズムの同定を試みています。